ISO 10993‑7:2026: Addressing Patient Safety Concerns Around EO Residuals
Read Blog

Are Your Medical Devices Categorized Correctly? Key Insights from the Proposed ISO 10993-1 Revision
Read Blog

PFAS Compliance and Medical Device Innovation
Discover how PFAS regulations impact medical device manufacturing, exploring the balance between unique material properties and safety compliance.
Read Article

CXOToday Interview on PFAS Regulations
Explore how evolving PFAS regulations are driving innovation in the medical device industry, prompting a shift towards safer material alternatives without compromising device quality.
Read ArticleEnsuring Medical Device Safety and Reliability with Biocompatibility Evaluations
As medical devices become more advanced and integrated into patient care, biocompatibility has emerged as a critical focus area. Regulatory bodies are tightening oversight, and product recalls due to material-related safety concerns. In this environment, manufacturers must go beyond checkbox compliance, adopting a strategic, risk-based approach that ensures patient safety, meets global regulatory expectations, and supports timely market entry without inflating development costs.
At Tata Elxsi, we offer comprehensive medical device biocompatibility evaluation services across device lifecycle, backed by a multidisciplinary team of experts in toxicology, material science, clinical research, and regulatory affairs. With all capabilities under one roof, we efficiently manage large portfolios, scale quickly, and adapt to your needs, delivering consistent, compliant, and ethical solutions. Our proven methodologies cater to global requirements and our risk-based biocompatibility testing strategies reduce costs and accelerate time-to-market. With strategic thought leadership and agile delivery models, we empower medical device manufacturers to meet evolving standards with confidence ensuring safe, compliant, and high-quality devices that improve patient outcomes.
Ensuring Excellence in Biocompatibility Compliance as per the new EU MDR Standards




Here’s How We Can Help
Accelerating Biological Evaluation & Toxicology Through AI
- AI-powered automation for Biological Evaluation Reports (BERs) and toxicological risk assessments.
- Automated data aggregation, literature review, and evidence extraction to reduce manual effort.
- Rules-based drafting with Expert Human in the Loop (HITL) to ensure scientific accuracy and regulatory defensibility.
Smarter Risk Identification & Assessment
- Intelligent risk signal identification using deterministic logic and advanced analytics.
- Automation-enabled support for chemicals data enablement, hazard identification, exposure assessment, and risk characterization.
- Consistent, traceable, and data-driven toxicology workflows.
Faster, Scalable, and Regulatory-Ready
- Streamlined assessment workflows that significantly reduce project timelines.
- Improved data quality, consistency, and decision-making confidence.
- Scalable solutions supporting medical device biocompatibility and global regulatory compliance.
Medical Device Biocompatibility Service Offerings
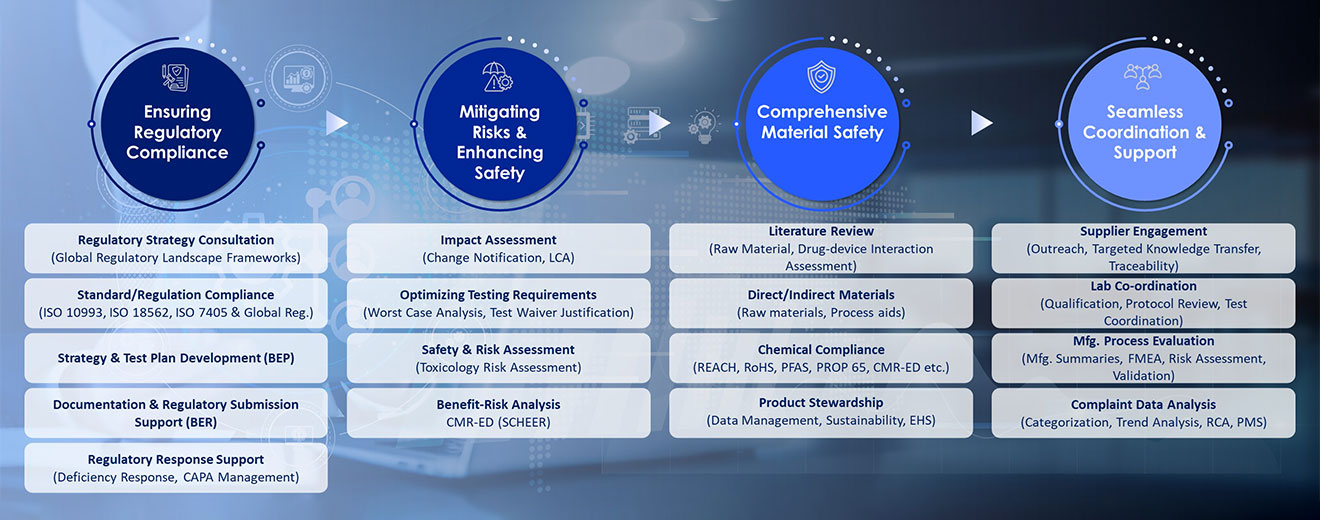
Our biocompatibility framework delivers end-to-end support for regulatory-compliant product safety evaluation. We integrate with the evolving regulatory trends and follow a risk-based framework, coupled with robust data gathering, supplier coordination and documentation strategies to streamline regulatory submissions and minimize delays. Our approach ensures your devices meet global safety standards throughout their lifecycle. With deep insights into the evolving global regulatory frameworks, we enable harmonized compliance strategies to ISO 10993, ISO 18562, ISO 7405 & global regulations that reduce duplication and accelerate market access.
Strategy Development and Lifecycle Solutions
- End-to-end strategies for NPI, legacy devices, M&A, and lifecycle and change management (LCM).
- Tailored pathways to navigate complex regulations with global harmonization expertise.
- Integration of biological safety across the product lifecycle, from concept to post-market.
Regulatory Intelligence and Proactive Compliance
- BEP/ BERs with risk-based frameworks aligned with current and emerging global standards.
- Strategic insights to anticipate regulatory shifts and market challenges.
- Re-evaluations, alternative material strategies, TRA and proactive risk mitigation.
Sustainability and Chemical Compliance
- Compliance with global chemical regulations including REACH, RoHS, TSCA, PFAS, and sustainabiliy.
- BoM screening, supplier engagement, and declaration audits.
- Risk-benefit assessments of CMR EDs (SCHEER guidelines) and substances of concern.
Why Tata Elxsi
- Regulatory excellence with advanced scientific expertise and evidence-based medical device biocompatibility strategies.
- Scalable programs with early risk detection, strategic test planning, and strong justifications to accelerate innovation.
- Efficient frameworks using worst-case modeling to minimize redundant biocompatibility testing and optimize resources.
- Tailored solutions for complex, high-risk devices with predictive scheduling to maximize lab throughput.
- Strategic risk-based methodologies leveraging chemical data and toxicological literature for compliance.
In Focus


Information Hub
-
What are global regulatory expectations for biocompatibility, and how does ISO 10993 compliance align with these requirements?
Regulators require risk-based evaluations based on device use, contact type, and duration. ISO 10993 provides the core framework, while markets differ in data needs, submission formats, and in the treatment of chemical characterization and substances of concern.
-
How does Toxicological Risk Assessment (TRA) fit into the overall biocompatibility evaluation?
TRA is more than a regulatory requirement, it safeguards patient safety. While biological tests show how a device behaves in the body, TRA uncovers hidden chemical risks that could impact patient safety over time. It’s a regulatory must-have under ISO 10993-1 and a key to building faster, stronger submissions to the FDA and EU Notified Bodies.
-
How should biocompatibility assessment account for device life cycle changes?
Biocompatibility assessment must be carried out throughout the device lifecycle, including design, manufacturing, use, aging, and reprocessing. A risk-based approach should validate existing data and use literature or supplier information where possible, with targeted testing for extractables, leachables, or material changes conducted when gaps remain.