Introduction
As the medical device landscape evolves, so do the standards that govern it. The industry is currently focused on the draft version of ISO/DIS 10993-1:2024, which addresses the biological evaluation of medical devices. This draft emphasizes integrating biological evaluation into a comprehensive risk management process, involving meticulous risk identification and assessment before testing.
Ensuring patient safety remains paramount, particularly through categorizing medical devices based on contact type and exposure duration. So, what exciting new clarifications and enhancements does the proposed revision bring for calculating contact duration and cumulative use? Dive into this blog to uncover the key changes and their implications for manufacturers, including:
Key changes related to device contact duration and category include:
- Streamlined device contact categories
- Revised Definitions for contact duration, including intermittent contact.
- Clarified exposure duration
Proposed ISO 10993-1 Revision - What You Need to Know!
The proposed revisions to ISO 10993-1 will redefine the biocompatibility evaluation process for medical devices. These changes, which clarify contact duration calculations and refine cumulative use guidelines, aim to streamline processes and significantly enhance patient safety. Let's explore the key updates and their profound implications for the industry.
- Refined Contact Categories
The categorization of medical devices has undergone a significant overhaul to provide clearer guidelines based on contact type. Exposure duration and contact types have been clarified, with the term 'externally communicating devices’ being removed. New categories based on contact type have been introduced, and the previous single table in Annex A of the 2018 standard has been divided into four distinct tables, each tailored to the specific type of contact for medical devices.
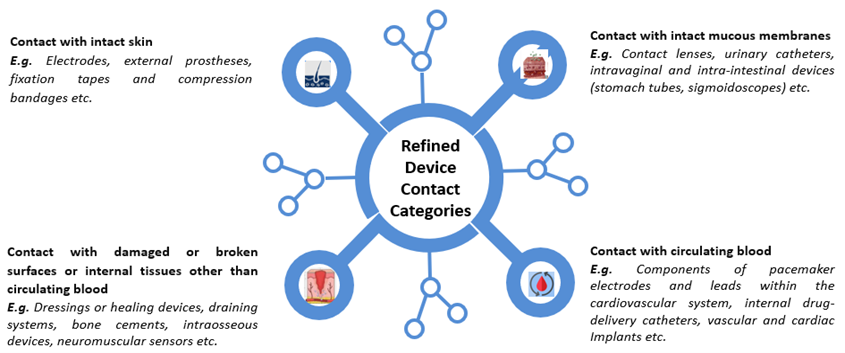 Figure 1.1: Depiction of Redefined Device Contact Categories
Figure 1.1: Depiction of Redefined Device Contact Categories
- Updates on Contact Duration and Cumulative Use
The ISO 10993-1:2018 standard defines contact duration as the cumulative sum of single, multiple, or repeated durations of contact. The draft version (ISO/DIS 10993-1:2024) maintains these considerations and provides further clarifications on calculating exposure duration for medical devices in daily contact with tissues and those in intermittent contact (with more than 24 hours between uses).
Another proposed revision aims to clarify cumulative use calculations. Discrepancies between manufacturers and regulatory bodies have complicated regulatory submissions. As per the proposed revision, when calculating the total exposure period, the period (in days) between the first and last use of a medical device should be considered. Examples include wound dressings, where total exposure is the complete duration of wound site treatment. However, for devices used briefly over many days (e.g., 10 minutes, twice a week for 6 months), counting the duration between the first and last day of treatment as total exposure may be an overestimation.
So, how do you navigate the complexities of calculating cumulative use for medical devices, especially when discrepancies arise between manufacturers and regulatory bodies? Let’s understand this with a real-world example.
Example: Disposable Feeding Tube
A patient recovering from surgery requires a disposable feeding tube for nutritional support, used for 1 hour daily over a 10-day period. According to the 2018 ISO 10993-1 standard, this device was classified as having limited contact with mucosal membranes. However, with the proposed revision, it is now reclassified as a prolonged contact device due to the cumulative contact days
This reclassification is more than a technicality—it significantly affects the required biological endpoint testing. Beyond the standard tests for cytotoxicity, sensitization, and irritation for limited-duration contact devices, manufacturers must now also consider biological testing for acute, subacute, and genotoxicity endpoints for prolonged contact devices to ensure device safety.
This example highlights the importance of accurate device categorization and the need for comprehensive testing to ensure patient safety. Manufacturers must ensure that the materials used are safe for the device use duration and that relevant testing is performed to meet the highest standard of care.
Updated Contact Duration: What's Next for Device Manufacturers?
With the proposed ISO 10993-1 revision, the categorization of medical devices based on exposure duration would bring several important implications, including:
- Device Recategorization: Devices may be recategorized based on cumulative use, shifting from limited to prolonged or prolonged to long-term contact duration.
- Safety and Biocompatibility: Devices with longer exposure must meet stricter biocompatibility requirements, including rigorous toxicological evaluation and testing.
- Regulatory Compliance: Manufacturers must adhere to specific regulatory requirements based on exposure duration to ensure safety and compliance with international standards.
- Design and Material Selection: Categorization impacts material and design choices; long-term exposure devices may require more durable and degradation-resistant materials.
These changes underscore the need for manufacturers to stay updated with the latest standards to ensure the safety and effectiveness of their medical devices.
Risk Mitigation - Addressing Manufacturer Concerns
As medical device manufacturers adapt to the proposed ISO 10993-1 revision, they may have questions about its impact on current practices and documentation. The following Q&A section addresses some key concerns and offers steps for compliance.
Q1: How will the revision impact the current biological evaluation documents?
A1: The revision may require updates to current biological evaluation documents. Conducting a gap analysis will help identify necessary updates and ensure documentation aligns with the new standards.
Q2: Why is it important to review current documentation?
A2: Reviewing current documentation through a gap analysis helps identify necessary updates in biological evaluation documents, ensuring compliance with the latest standards.
Q3: Are there additional aspects that require evaluation?
A3: Yes, manufacturers may need to reassess device categorization based on refined contact categories and updated exposure duration calculations to ensure accurate evaluations.
Q4: How can manufacturers perform effective risk analysis and mitigation?
A4: By conducting a detailed risk analysis, manufacturers can identify potential biological hazards such as cytotoxicity, sensitization, irritation, genotoxicity, and carcinogenicity, and develop strategies to mitigate these risks.
Q5: How should manufacturers handle remediating documentation?
A5: Updating the Biological Evaluation Report (BER) with detailed justifications for biological risk assessments and ensuring transparency in decision-making is crucial for compliance and clarity.
Q6: How should manufacturers prepare for changes related to device contact duration and category?
A6: To get ready for these changes, manufacturers can start with the following steps:
- Review Current Documentation: Conduct a gap analysis to identify updates needed in biological evaluation documents.
- Identify Additional Evaluation Aspects: Reassess device categorization based on refined contact categories and updated exposure duration calculations.
- Risk Analysis and Mitigation: Perform a detailed risk analysis to identify potential biological hazards such as cytotoxicity, sensitization, irritation, genotoxicity, and carcinogenicity.
- Biological Evaluation Plan (BEP): Update BEP to align with new requirements and integrate it into the overall risk management process.
- Testing: Plan for any additional testing required by the new standards.
- Remediating Documentation: Update the Biological Evaluation Report (BER) with detailed justifications for biological risk assessments and ensure transparency in decision-making.
By taking these proactive steps, manufacturers can ensure compliance, thereby meeting the latest standards and maintaining the highest level of patient safety.
Conclusion
As the medical device industry evolves, staying ahead of regulatory changes is crucial. The proposed ISO 10993-1 revision emphasizes integrating biological evaluation into risk management, aligning with ISO 14971. This shift to a risk-based approach can streamline compliance and reduce redundant testing. Embracing these updates will help manufacturers streamline processes and reinforce patient safety.
Now is the time for manufacturers to review changes, update practices, and engage with regulatory bodies. Consulting experts can save costs and time for testing and submissions. By adapting proactively, you contribute to a safer, more effective healthcare environment. Stay informed, stay prepared, and lead in medical device innovation.
Author
Bio-compatibility & Toxicology Subject Matter Expert
Tata Elxsi